Installation and running¶
MAGERI is distributed in a form of executable JAR file and requires Java v1.8 to run. Source code and binaries can be found in corresponding repository. The latest release can be found here. MAGERI can be executed by running
java -Xmx32G -jar mageri.jar [arguments]
The -Xmx32G option sets the memory usage limit that should be enough
for most datasets. The list of arguments is given by -h option
and described below as well.
| option | argument | description |
|---|---|---|
-R1 |
fastq[.gz] | First read file. |
-R2 |
fastq[.gz] | Second read file [optional]. |
--platform |
string | Platform: Illumina, IonTorrent or Roche454 [default = Illumina]. |
--library-type |
string | Library prep start, SS (single-stranded, linear PCR) or DS (double-stranded) [default=SS]. |
--project-name |
string | Project name [optional]. |
--sample-name |
string | Sample name [optional]. |
-O |
path | Path to output folder [default = current folder]. |
-M1 |
metadata file | De-multiplexing, primer trimming and barcode extraction using specified metadata file. See Multiplex (M1) |
-M2 |
metadata file | Primer trimming and barcode extraction using specified metadata file. See Primer (M2) |
-M3 |
mask1[:mask2] | Positional UMI extraction. See Positional (M3) |
-M4 |
Preprocessed data, UMI information is stored in header. See Header (M4) | |
--references |
FASTA file | File with reference sequences. See Sequences |
--bed |
BED file | Genomic coordinates of reference sequences [optional]. See Coordinates |
--contigs |
metadata file | File with contig names, lengths, and assembly id [optional]. See Contigs |
Important
One of M1-4 options should be specified. While
--bed and --contigs parameters are optional, if not specified,
resulting SAM and VCF files could not be directly annotated and
visualized using data from corresponding genome assembly.
Input¶
MAGERI accepts paired or single-end raw sequencing reads in FASTQ format with Phred33 quality encoding, either uncompressed or GZIP-compressed. To run the pipeline, user should specify pre-processing options which tell the software how to extract UMI sequences and sample barcodes in case the latter are incorporated into the adapter sequence. Additionally, genomic information for reference sequences can be provided to be used during mapping and VCF/SAM file generation.
Note
Genomic information bundle for genes from Cancer Gene Census (hg38 genome assembly) is available here. This bundle contains all exons with +/-50 base flanks. Additional instructions to create genomic information bundle for your own list of genes are given below.
Pre-processing¶
Four de-multiplexing modes are available in MAGERI to handle the majority of possible library designs.
Multiplex (M1)¶
In this mode it is assumed that samples are multiplex with barcodes in adapter sequences that additionally cary a UMI tag. In this case tab-delimited barcodes table should be provided, e.g.:
| sample_name | master_first | master_adapter | slave_adapter |
|---|---|---|---|
| Sample1 | 0 | NNNNNNNNNNNNNNctctcATGC | GATTTttcaNNNNNNNNNNNNNN |
| Sample2 | 1 | NNNNNNNNNNNNNNtgaaATAGC | GCATgagagaNNNNNNNNNNNNNN |
| Sample3 | 1 | NNNNNNNNNNNNNNtgaaTAGCA | GCATgagagaNNNNNNNNNNNNNN |
| Sample4 | 1 | NNNNNNNNNNNNNNtgaaATAGC | |
| ... |
Samples are de-multiplexed based on master adapter sequence and filtered for matching slave adapter sequence in case it is provided (paired-end data). Master adapter is first searched in both read#1 and read#2. Then the mate read of the master adapter-containing read is reverse-complemented and searched for slave adapter sequence if it is provided. Simply speaking, master and slave adapter sequences should be provided as if they were on the same strand.
After matching and UMI extraction, reads are oriented to be on the same strand and adapter sequences are trimmed.
If master_first is set to 0 reads are swapped and reverse-complemented.
De-multiplexed samples are further analyzed separately.
The following rules apply to master and slave adapter sequence specification:
- Slave adapter sequence could be omitted, master adapter sequence should be unique for each sample.
- Adaptor sequence can contain any IUPAC DNA letters.
- Upper and lower case letters mark seed (exact match) and fuzzy-search region parts respectively.
- N characters mark UMI region to be extracted.
- Multiple rows could correspond to the same sample
Primer (M2)¶
This is a variant of -M1 mode that extracts UMIs and removes primer sequences, but processes
all reads together as if they were coming from the same sample, e.g.
| region_name | master_first | left_primer | right_primer |
|---|---|---|---|
| ARAF_E7_F | 1 | NNNNNNNNNNNNNNactgtGACCCGGAgcact | cacaGGGCAGAGggtagag |
| BRAF_E15_F | 1 | NNNNNNNNNNNNNNcataaTGCTTGCTctgatagga | ggagTGGGTCCCatcagttt |
| ... |
Positional (M3)¶
This mode is specified by one or two masks which are used to scan the first read and the reverse complement of the second read. As always, N characters are used to specify UMI positions. Nucleotides (including ambiguity codes) require exact match, while n characters are used to specify offset. For example
-M3 NNNNNwill use first 5 bases of read#1 as UMI.-M3 nnnNNNNNwill use bases from 4 to 8 of read#1 as UMI.-M3 nnnNNNNNatgcwill scan read#1 for nnnNNNNNatgc, nnNNNNNatgc, nNNNNNatgc and NNNNNatgc until matching the atgc string.-M3 NNNNN:NNNNNwill extract first 5 bases of read#1 and last 5 bases of reverse complement of read#2.
Warning
This mode should be used with care for non-oriented reads, as only one read pair orientation will be scanned.
Header (M4)¶
If this mode is specified, it is assumed that FASTQ files contain UMI:NNN:QQQ entry in read headers, separated by tab or space from other header entries. Here NNN are UMI nucleotides and QQQ are corresponding quality Phred scores.
Note
In case working with a large set of primers/adapters, it is common to misspecify several of them.
It is advised to first manually check for primer extraction efficiency and troubleshoot incorrect ones.
To do so for both M1 and M2 cases, run MAGERI in M1 mode and tell it to
take only a fraction of reads, say 10000, with --limit 10000 and inspect resulting *.checkout.txt
output file to see if any of the primer sequences were not extracted. To figure out real primer sequences
one can run in the M3 mode specifying only UMI positions and then check resulting SAM files in IGV to
get sequences of corresponding regions. Those sequences can then be manually checked against the primer set
to correct errors in primer sequences.
Genomic information¶
Sequences¶
MAGERI requires a reference file in FASTA format to run the alignment and variant calling.
Note that by default more than 30% of MIG consensus sequence should align to targeted region,
so ideally adapter/primer trimming is recommended. In case of targeted capture (e.g. exome sequencing),
upstream and downstream regions (+/- readlength/2 bases) of exons should be included.
Typical reference FASTA file should look like
>HER3_E2
CGGCGATGCTGAGAACCAATACCAGACACTGTACAAGCTCTACGAGAGGTGTGAGGTGGTGATGGGGAACCTTGAGATTGTGCTCACGGGAC
>HER3_E3
CTATGTCCTCGTGGCCATGAATGAATTCTCTACTCTACCATTGCCCAACCTCCGCGTGGTGCGAGGGACCCAGGTCTACGATGGGAAGTTTGCCATCTTCGTCATGTTGAACTATAACACCAACTCC
>HER3_E6
TTCTCTCCTTCCATAGTGACCAAGACCATCTGTGCTCCTCAGTGTAATGGTCACTGCTTTGGGCCCAACCCCAACCAGTGCTGCCATGATGAGTGTGCCGGGGGCTGCTCAGGCCCTCAGGACACAGACTGCTTTGTATG
>HER3_E7
CCACAGCCTCTTGTCTACAACAAGCTAACTTTCCAGCTGGAACCCAATCCCCACACCAAGTATCAGTATGGAGGAGTTTGTGTAGCCAGCTGTCCCCGTAAGTGTCTGAGGGGAAGGA
>HER3_E8
TCATCTCTAATGGTGTCCTCCTCCTCTTCCCTAGATAACTTTGTGGTGGATCAAACATCCTGTGTCAGGGCCTGTCCTCCTGACAAGATGGAAGTAGATAAAAATGGGCTCAAGATGTGTGAGCCTTGTGGGGGACTATGTCCCAAAGGTGGGTAG
>HER3_E9
GGGAACAGGCTCTGGGAGCCGCTTCCAGACTGTGGACTCGAGCAACATTGATGGATTTGTGAACTGCACCAAGATCCTGGGCAACCTGGACTTTCTGATCAC
>HER3_E21
TACAGGGAATGTACTACCTTGAGGAACATGGTATGGTGCATAGAAACCTGGCTGCCCGAAACGTGCTACTCAAGTCACCCAGTCAGGTTCAGGTGGCAGATTTTGGTGTGGCTGACCTGCTGCCTCCTGATGATAAGCAGC
>HER3_E23
TTCCTGCAACAGGTGTGACAGTTTGGGAGTTGATGACCTTCGGGGCAGAGCCCTATGCAGGGCTACGATTGGCTGAAGTACCAGACCTGCTAGAGAAGGGGGAGCGGTTGGCACAGCCCCAGATCTGCACAATTGATGTCTACA
Coordinates¶
In order to make output feasible for post-analysis and visualization, a BED file containing genomic coordinates of references should be included. For the example FASTA file above it should be
#chr start end name unused strand
chr12 56477568 56477659 HER3_E2 0 +
chr12 56478798 56478924 HER3_E3 0 +
chr12 56481562 56481701 HER3_E6 0 +
chr12 56481849 56481966 HER3_E7 0 +
chr12 56482292 56482447 HER3_E8 0 +
chr12 56482538 56482639 HER3_E9 0 +
chr12 56491563 56491703 HER3_E21 0 +
chr12 56492530 56492673 HER3_E23 0 +
Note
FASTA entries that do not have corresponding BED rows will be skipped from SAM and VCF output.
Note
The most straightforward way (in my experience) to generate FASTA and BED files is
to use ENSEMBL Biomart. Specify your
gene identifiers in the Filters section and choose Sequences as output mode.
Don’t forget to manually add flanking bases count (in case you specify them) to BED file
as they’re not accounted for in Biomart output. Importantly, ENSEMBL coordinates are 1-based,
while BED format is 0-based, so adjust appropriately by subtracting 1 from start coordinate in
BED.
A step-by-step instruction on getting your references from Ensembl BioMart is given below:
- Go to Martview.
- In the dataset section select Ensembl genes 84 and choose Homo sapiens genes.
- In the filter section and Gene subsection, input your gene IDs to external references ID textbox and select appropriate nomenclature (e.g. HGNC symbol when using gene symbols).
- In attributes section select Sequences and untick all header information features.
- Under the SEQUENCES menu select Exon sequences and input the 5’ and 3’ flank base count, e.g. 50 (it should be the same for both 5’ and 3’ flank). This is to ensure that reads produced by exome capture techniques are fully mapped.
- Select the following features (order matters here): Associated Gene Name, Ensembl Exon ID, Chromosome Name, Exon Chr Start (bp), Exon Chr End (bp) and Strand.
- Go to results, select unique results only and download the resulting FASTA file (save it as
biomart_refs.fa). - Download and run the following script (requires Groovy to be installed):
groovy ExtractBedFormBiomartRefs.groovy biomart_refs.fa refs.fa refs.bed 50. The last argument specifies the flank size. - You can now supply resulting
refs.fa,refs.bedand this contigs file when running the pipeline.
Contigs¶
Genome assembly metadata file is required to create SAM and VCF file headers, here is an example tab-delimited table for hg19 genome assembly
#chrom assembly length
chr12 hg19 133851895
Again, contig names (chr12,...) and coordinates in BED file should be concordant with assembly metadata file.
Note
If assembly for a given contig is named PRIVATE, corresponding results will be skipped SAM and VCF output (but not from internal MAGERI output files).
Output¶
MAGERI generates multiple internal output files summarizing the results of each pipeline step:
*.checkout.txt- de-multiplexing and UMI extraction yield*.umi.histogram.txt- MIG size distribution*.assemble.txt- MIG consensus assembly efficiency;*.assemble.R1/2.fastq.gz- assembled consensus sequences in FASTQ format with CQS quality scores*.mapper.txt- MIG consensus mapping statistics for each reference*.variant.caller.txt- tab-delimited file with variant calls (in original reference coordinates, not genomic ones)
Those files are useful for analysis quality control, for example, *.checkout.txt should be monitored to
ensure correct primer specificaiton and *.umi.histogram.txt should contain a clear peak that can be
thresholded with 5+ reads per UMI to check if library prep yields optimal starting molecule coverage.
Additionally, mapping and variant calling results are provided in SAM and VCF formats
Example SAM output:
@HD VN:1.0 SO:unsorted GO:query
@SQ SN:chr1 LN:249250621 AS:hg19
@SQ SN:chr2 LN:243199373 AS:hg19
@SQ SN:chr3 LN:198022430 AS:hg19
@SQ SN:chr4 LN:191154276 AS:hg19
@SQ SN:chr5 LN:180915260 AS:hg19
@SQ SN:chr6 LN:171115067 AS:hg19
@SQ SN:chr7 LN:159138663 AS:hg19
@SQ SN:chr8 LN:146364022 AS:hg19
@SQ SN:chr9 LN:141213431 AS:hg19
@SQ SN:chr10 LN:135534747 AS:hg19
@SQ SN:chr11 LN:135006516 AS:hg19
@SQ SN:chr12 LN:133851895 AS:hg19
@SQ SN:chr13 LN:115169878 AS:hg19
@SQ SN:chr14 LN:107349540 AS:hg19
@SQ SN:chr15 LN:102531392 AS:hg19
@SQ SN:chr16 LN:90354753 AS:hg19
@SQ SN:chr17 LN:81195210 AS:hg19
@SQ SN:chr18 LN:78077248 AS:hg19
@SQ SN:chr19 LN:59128983 AS:hg19
@SQ SN:chr20 LN:63025520 AS:hg19
@SQ SN:chr21 LN:48129895 AS:hg19
@SQ SN:chr22 LN:51304566 AS:hg19
@SQ SN:chrX LN:155270560 AS:hg19
@SQ SN:chrY LN:59373566 AS:hg19
@RG ID:3 SM:h1-1 PU:h1-1 LB:p126-1 PL:ILLUMINA
@PG ID:mageri VN:1.0.0 CL:mageri-1.0.0.jar -I project-1.json -O output/
TGTATATCCCCTGA 16 chr1 115258663 30 20S131M22S * 0 0 AGGTCAGCGGGCTACCACTGGGCCTCACCTCTATGGTGGGATCATATTCATCTACAAAGTGGTTCTGGATTAGCTGGATTGTCAGTGCGCTTTTCCCAACACCACCTGCTCCAACCACCACCAGTTTGTACTCAGTCATTTCACACCAGCAAGAACCTGTTGGAAACCAGTAA GHGGHHHHHHHHHHHHHHHHHHHHHHHHIHIHHHHHHHHHHHHHIHHHHHIHIHIHHIHHHHHIIIIHHIHHHHIHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHIHHHHHIHHHHHHHHHHHHHHHHHHHHHHHHHHHHHIHHIIHHHIHHIHHHHHHHHHIIHIHHHHIH RG:Z:3
GTGTAATTAAATGA 0 chr2 209113093 28 22S103M21S * 0 0 CATTATTGCCAACATGACTTACTTGATCCCCATAAGCATGACGACCTATGATGATAGGTTTTACCCATCCACTCACAAGCCGGGGGATATTTTTGCAGATAATGGCTTCTCTGAAGACCGTGCCACCCAGAATATTTCGTATGGTG HHHIHHHHHIHIHIIHHHIIHIIHIHIIHHHGIIIHIHHHHIHHHHHHHHHHHHHHIHHIIIHHHHHIHHHHHHHIHHHHHHHHHHHIIIHIIHHIHHHHHHHHHHHHHHHHHIHHHIHHHHHIHIHHHHHHHHHHHHHHHHHHHH RG:Z:3
Example VCF output:
##fileformat=VCFv4.0
##fileDate=Tue Jun 02 05:30:36 GMT+03:00 2015
##source=mageri-1.0.0
##reference=file:///data/misha/P126/meta/refs.fa
##contig=<ID=chr1,assembly=hg19,length=249250621>
##contig=<ID=chr2,assembly=hg19,length=243199373>
##contig=<ID=chr3,assembly=hg19,length=198022430>
##contig=<ID=chr4,assembly=hg19,length=191154276>
##contig=<ID=chr5,assembly=hg19,length=180915260>
##contig=<ID=chr6,assembly=hg19,length=171115067>
##contig=<ID=chr7,assembly=hg19,length=159138663>
##contig=<ID=chr8,assembly=hg19,length=146364022>
##contig=<ID=chr9,assembly=hg19,length=141213431>
##contig=<ID=chr10,assembly=hg19,length=135534747>
##contig=<ID=chr11,assembly=hg19,length=135006516>
##contig=<ID=chr12,assembly=hg19,length=133851895>
##contig=<ID=chr13,assembly=hg19,length=115169878>
##contig=<ID=chr14,assembly=hg19,length=107349540>
##contig=<ID=chr15,assembly=hg19,length=102531392>
##contig=<ID=chr16,assembly=hg19,length=90354753>
##contig=<ID=chr17,assembly=hg19,length=81195210>
##contig=<ID=chr18,assembly=hg19,length=78077248>
##contig=<ID=chr19,assembly=hg19,length=59128983>
##contig=<ID=chr20,assembly=hg19,length=63025520>
##contig=<ID=chr21,assembly=hg19,length=48129895>
##contig=<ID=chr22,assembly=hg19,length=51304566>
##contig=<ID=chrX,assembly=hg19,length=155270560>
##contig=<ID=chrY,assembly=hg19,length=59373566>
##phasing=none
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency">
##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele">
##INFO=<ID=CQ,Number=1,Type=Integer,Description="Assembly quality">
##FILTER=<ID=q20,Description="Quality below 20">
##FILTER=<ID=si10000,Description="Singleton, frequency below 10000">
##FILTER=<ID=c100,Description="Coverage below 100">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##INFO=<ID=DP,Number=1,Type=Integer,Description="MIG Depth">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT p126-1.h1-1
chr1 115252206 . G A 16 q20 DP=307;AF=0.0032573289;AA=G;CQ=39.0 GT:DP 0/1:307
chr1 115258758 . C T 383 . DP=542;AF=0.040590405;AA=C;CQ=39.0 GT:DP 0/1:542
Those files can be further used in downstream analysis. For example, SAM files can be viewed in IGV browser, while VCF files can be annotated with SnpEff. It is always recommended to inspect variant calls and alignment data in IGV to ensure there are no alignment artefacts.
Example¶
A toy example dataset can be downloaded from here. Unpack it and run the following command:
java -jar mageri.jar -M2 primers.txt --references refs.fa -R1 example_R1.fastq.gz -R2 example_R2.fastq.gz out/
The resulting VCF file should contain 31:T>C, 88:T>C and 89:T>C variants. Next, SAM files
can be converted to BAM format, sorted and indexed using
samtools. Indexed BAM files can be browsed in Integrative Genome Viewer
using refs.fa as user-provided genome. Manual inspection of alignments should reveal that mutations
at positions 31 and 88 are linked:
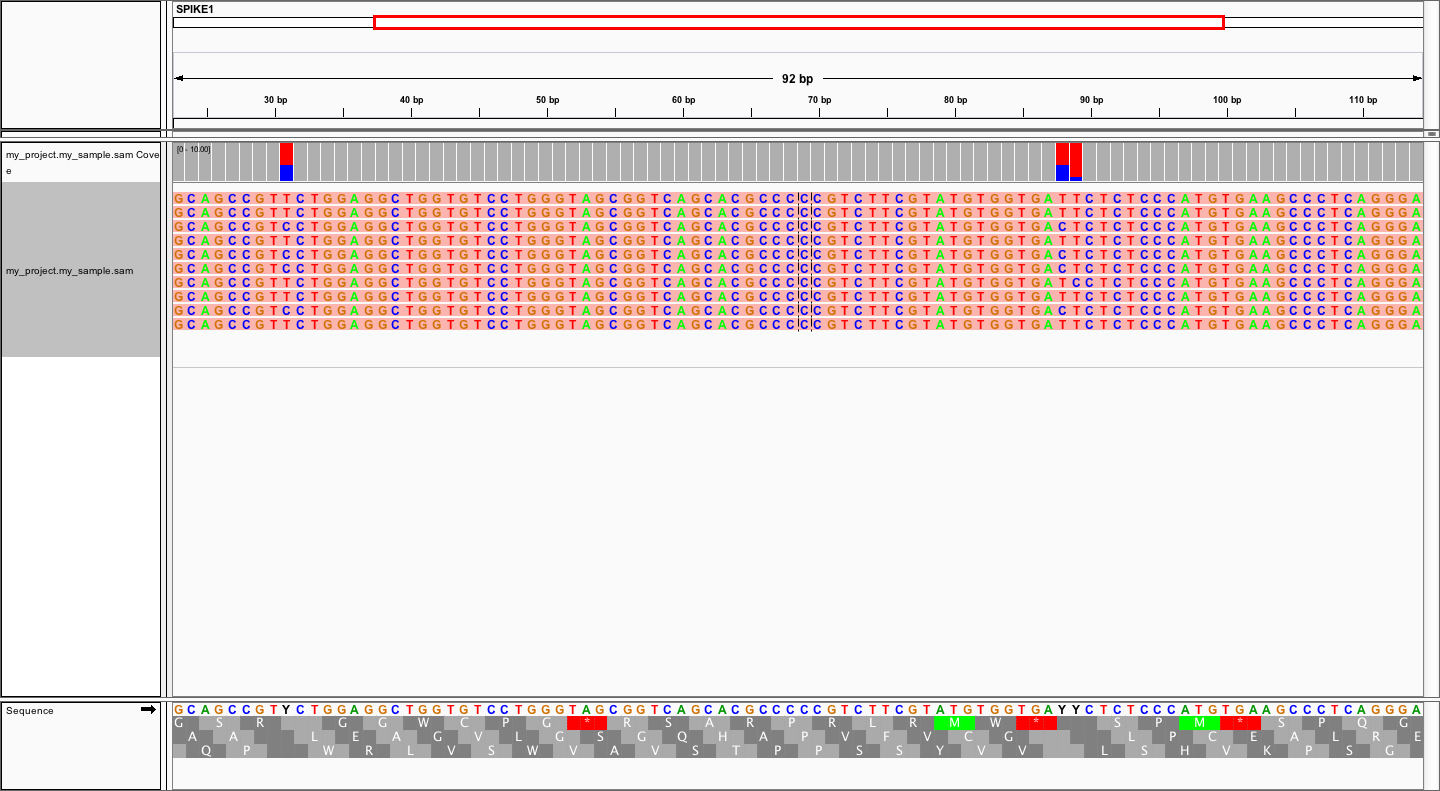
In order to evaluate MAGERI software and learn its capabilities I recommend checking out the mageri-paper repository containing real-world datasets, metadata and shell scripts that can be used to run MAGERI analysis.
Advanced¶
Presets¶
Importing and exporting parameters¶
By default MAGERI runs with pre-configured and optimized parameters, so change them only if you know what you are doing. The parameter config can be changed by exporting, modifying and re-importing corresponding XML file:
java -jar mageri.jar --export-preset my_preset.xml
gedit my_preset.xml
...
java -Xmx64G -jar mageri.jar --import-preset my_preset.xml [arguments]
Note that the --platform and --library-type arguments can be specified to alter the
default preset. The content of the default XML config file is given below:
<?xml version="1.0" encoding="UTF-8"?>
<MageriPresets>
<version>1.0.1-SNAPSHOT</version>
<platform>ILLUMINA</platform>
<libraryType>SS</libraryType>
<DemultiplexParameters>
<orientedReads>false</orientedReads>
<maxTruncations>2</maxTruncations>
<maxGoodQualMMRatio>0.05</maxGoodQualMMRatio>
<maxLowQualityMMRatio>0.1</maxLowQualityMMRatio>
<lowQualityThreshold>20</lowQualityThreshold>
</DemultiplexParameters>
<PreprocessorParameters>
<umiQualThreshold>10</umiQualThreshold>
<goodQualityThreshold>30</goodQualityThreshold>
<trimAdapters>true</trimAdapters>
<minUmiMismatchRatio>20.0</minUmiMismatchRatio>
<forceOverseq>false</forceOverseq>
<defaultOverseq>5</defaultOverseq>
</PreprocessorParameters>
<AssemblerParameters>
<offsetRange>4</offsetRange>
<anchorRegion>8</anchorRegion>
<maxMMs>4</maxMMs>
<maxConsequentMMs>0</maxConsequentMMs>
<maxDroppedReadsRatio>0.3</maxDroppedReadsRatio>
<maxDroppedReadsRatioAfterRescue>0.0</maxDroppedReadsRatioAfterRescue>
<maxTrimmedConsensusBasesRatio>0.3</maxTrimmedConsensusBasesRatio>
<minMatchedBasesInRealignedReadRatio>0.0</minMatchedBasesInRealignedReadRatio>
<pcrMinorTestPValue>0.001</pcrMinorTestPValue>
<cqsRescue>false</cqsRescue>
<qualityTrimming>true</qualityTrimming>
<greedyExtend>true</greedyExtend>
</AssemblerParameters>
<ReferenceLibraryParameters>
<splitLargeReferences>true</splitLargeReferences>
<maxReferenceLength>1000</maxReferenceLength>
<readLength>100</readLength>
</ReferenceLibraryParameters>
<ConsensusAlignerParameters>
<k>11</k>
<matchReward>1</matchReward>
<mismatchPenalty>-3</mismatchPenalty>
<gapOpenPenalty>-6</gapOpenPenalty>
<gapExtendPenalty>-1</gapExtendPenalty>
<minIdentityRatio>0.9</minIdentityRatio>
<minAlignedQueryRelativeSpan>0.7</minAlignedQueryRelativeSpan>
<muationCqsThreshold>30</muationCqsThreshold>
<useSpacedKmers>true</useSpacedKmers>
</ConsensusAlignerParameters>
<VariantCallerParameters>
<noIndels>false</noIndels>
<qualityThreshold>20</qualityThreshold>
<singletonFrequencyThreshold>10000</singletonFrequencyThreshold>
<coverageThreshold>100</coverageThreshold>
<errorModelType>MinorBased</errorModelType>
<modelOrder>1.0</modelOrder>
<modelCycles>20.0</modelCycles>
<modelEfficiency>1.8</modelEfficiency>
<modelCoverageThreshold>100</modelCoverageThreshold>
<modelMinorCountThreshold>10</modelMinorCountThreshold>
<substitutionErrorRateMatrix>0.0,1.0E-6,1.0E-6,1.0E-6;1.0E-6,0.0,1.0E-6,1.0E-6;1.0E-6,1.0E-6,0.0,1.0E-6;1.0E-6,1.0E-6,1.0E-6,0.0</substitutionErrorRateMatrix>
</VariantCallerParameters>
</MageriPresets>
Presets are also available for 454 and IonTorrent platforms that are characterized by indel errors at homopolymer regions and therefore require a more robust
algorithm for consensus assembly, and therefore different <AssemblerParameters> preset:
java -jar mageri.jar --platform roche454 --export-preset my_preset.xml
Will have the following difference:
...
<platform>ROCHE454</platform>
...
<AssemblerParameters>
<offsetRange>4</offsetRange>
<anchorRegion>8</anchorRegion>
<maxMMs>4</maxMMs>
<maxConsequentMMs>2</maxConsequentMMs>
<maxDroppedReadsRatio>0.7</maxDroppedReadsRatio>
<maxDroppedReadsRatioAfterRescue>0.3</maxDroppedReadsRatioAfterRescue>
<maxTrimmedConsensusBasesRatio>0.3</maxTrimmedConsensusBasesRatio>
<minMatchedBasesInRealignedReadRatio>0.5</minMatchedBasesInRealignedReadRatio>
<pcrMinorTestPValue>0.001</pcrMinorTestPValue>
<cqsRescue>true</cqsRescue>
<qualityTrimming>true</qualityTrimming>
<greedyExtend>false</greedyExtend>
</AssemblerParameters>
...
Parameter descriptions¶
Below goes the description of each parameter that can be changed in configuration XML file.
Preset
versionversion of software that generated this preset via--export-preset.platformname of the platform for which the preset was generated, specified with--platformoption during--export-preset. Allowed values areillumina,roche454andiontorrent. The preset affects consensus assembler parameters.libraryTypetype of library, single-stranded (SS) or double-stranded (DS), specified with--library-typeoption during--export-preset. This affects the consensus assembler and minor-based error model parameters.
De-multiplexing
orientedReadsif set tofalsewill search both read orientations for UMI inM1andM2cases, otherwise will search only the original readmaxTruncationsmaximum number of non-seed nucleotides that fall out the read boundaries (M1andM2mode)maxGoodQualMMRatiomaximum number of allowd non-seed mismatches with quality greater or equal tolowQualityThreshold(M1andM2mode)maxLowQualityMMRatiomaximum number of allowd non-seed mismatches with quality less thanlowQualityThreshold(M1andM2mode)lowQualityThresholdused in primer/adapter matching see above
Pre-processing
umiQualThresholdUMIs that have at least one base with quality less than that threshold will be discardedgoodQualityThresholdquality threshold used to mask nucleotides for minor-based error model (MBEM) used in variant callertrimAdaptersspecifies whether to trim found primer/adapter sequencesminUmiMismatchRatiominimum ratio of reads associated with parent and child UMI sequences, used to filter errors in UMI sequenceforceOverseqspecifies whether to enforcedefaultOverseqthreshold or to estimate one from MIG size histogramdefaultOverseqthreshold for number of reads in MIGs, used to filter unusable, erroneous and artefact UMIs
Consensus assembly
offsetRangeread offsets (from-offsetRangeto+offsetRange) to try when aligning readsanchorRegionhalfsize of region used to compare reads during alignemntmaxMMsmaximum number of mismatches inanchorRegion, reads having more thatmaxMMsmismatches in any offset will be droppedmaxConsequentMMsmaximum number of consequent mismatches between read and consensus during CQS rescue (seecqsRescuebelow). Reads that fail this filter are likely to contain an indel and are re-aligned to consensus using Smith-Waterman algorithm.maxDroppedReadsRatiomaximum ratio of reads dropped for a consensus to be discardedmaxDroppedReadsRatioAfterRescuemaximum ratio of reads dropped after CQS rescue (seecqsRescuebelow) for a consensus to be discardedmaxTrimmedConsensusBasesRatiomaximum ratio of bases trimmed from consensus due to poor CQS (seequalityTrimmingbelow) for a consensus to be discardedminMatchedBasesInRealignedReadRatiominimum fraction of matching bases during read re-alignment (seecqsRescuebelow) for a read to be droppedpcrMinorTestPValueP-value threshold used during PCR-induced minor error calling (seeminor calling)cqsRescueperform consensus quality score (CQS) rescue for indel-heavy readsqualityTrimmingtrim consensus bases with low consensus quality score which is proportional to the ratio of major base and total base countgreedyExtendspecifies whether to compute the initial PWM for maximal span of reads, uses average span if set tofalse
Reference library
splitLargeReferencessplit references larger thanmaxReferenceLengthinto partitions to speed up the consensus alignment and decrease its memory footprint.maxReferenceLengthmaximum length of reference, beyond which reference will be partitionedreadLengthestimate of max read length. In case reference is split, its paritions will contain overlapping regions to ensure that each read coming from a given reference will be fully contained in at least one of its paritions.
Consensus alignment
kk-mer size used by reference mapping algorithmmatchRewardmatch reward used by local alignment algorithmmismatchPenaltymismatch penalty used by local alignment algorithmgapOpenPenaltygap open penalty used by local alignment algorithmgapExtendPenaltygap extend penalty used by local alignment algorithmminIdentityRatiominimal local alignment identity (accounting for substitutions only) used for filteringminAlignedQueryRelativeSpanminimal relative span of query sequence that are aligned to reference, used for filteringmuationCqsThresholdconsensus quality threshold used to filter unreliable major mutationsuseSpacedKmersif set totruewill use k-mers with the central base set toN. This strategy (introduced in Vidjil software) can greatly improve mapping sensitivity while having the same specificity.
Variant calling
noIndelsif set totruewill not attempt to call indel variantsqualityThresholdvariant error model quality threshold, used in FILTER field of output VCF filesingletonFrequencyThresholdthreshold for ratio between signleton errors and their parent molecules (filters extremely rare errors introduced during UMI attachment), used in FILTER field of output VCF filecoverageThresholdthrehsold for variant coverage (number of MIGs), used in FILTER field of output VCF fileerrorModelTypeerror model type:MinorBasedinfer error rate from minor PCR errors that are deduced during consensus assembly (see Minor-Based Error Model aka MBEM),RawDatacompute error rate from average variant quality Phred score, orCustomthat uses error rates defined insubstitutionErrorRateMatrixmodelOrderorder of minor-based error model (MBEM), 1 for signle-stranded and 2 for double-stranded librarymodelCycleseffective number of PCR cycles used by MBEMmodelEfficiencyPCR efficiency value used by MBEMmodelCoverageThresholdcoverage threshold that is used in MBEM to decide whether to use error rate inferred at a given position or global error rate for a given substution type (e.g.A>G)modelMinorCountThresholdtotal number of inferred PCR minors at a given position that is used in MBEM to decide whether to use error rate inferred at a given position or global error rate for a given substution type (e.g.A>G)substitutionErrorRateMatrixa flat representation of substitution error rate matrix:0,A>G,A>C,A>T;G>A,0,G>C,G>T;C>A,C>G,0,C>T;T>A,T>G,T>C,0. Used ifCustomerror model is selected.
The parameters you are likely to change under certain conditions:
goodQualityThresholdin case reads are of poor sequencing quality (e.g. MiSeq 300+300 reads)readLengthwhen analyzing data generated by 454, IonTorrent platfroms and MiSeq long readsforceOverseqanddefaultOverseqin case MIG size histogram shows irregular behavior or5+reads per UMI coverage cannot be reachedmismatchPenalty,minIdentityRatioandminAlignedQueryRelativeSpanin case of a complex library and high number of artefact alignments; Note that a good solution to this problem is to introduce additional references (e.g. pseudogenes) if your reference set doesn’t cover everything that can be amplified with your primerserrorModelTypeandsubstitutionErrorRateMatrix.. we plan to publish a comprehensive set of error models inferred for different polymerases
Batch processing¶
MAGERI can be configured to run for multiple input files using a metadata config in JSON format. An example of metadata file is given below:
{
"project": "project_name",
"references": "metadata/refs.fa",
"bed": "metadata/refs.bed",
"contigs": "metadata/contigs.txt",
"structure": [
{
"byindex": [
{
"index": "group_name",
"r1": "R1.fastq.gz",
"r2": "R2.fastq.gz",
"submultiplex": {
"file": "metadata/adapters.txt"
}
}
]
},
{
"tabular": {
"file": "metadata/index1.txt",
"primer": {
"file": "metadata/primers.txt"
}
}
},
{
"tabular": {
"file": "metadata/index2.txt",
"positional": {
"mask1": "nnNNNNNNNNNNNN"
}
}
},
{
"tabular": {
"file": "metadata/index3.txt",
"preprocessed": {}
}
}
]
}
Here the byindex and tabular entries specify a sample group with corresponding FASTQ files
or index file, a tab-delimited table with sample_name\tfastq_R1\tfastq_R2 structure. The
submultiplex, primer, positional and preprocessed entries correspond to M1-4 demultiplexing
rules described above.
After the input.json and metadata/* files are prepared the entire pipeline can be run as follows:
java -Xmx32G -jar mageri.jar -I input.json -O output/
Example JSON files and metadata can be found here.